Amino Acid Workflows
Almost all Augur workflows describe nucleotide analyses. Here we describe workflows using entirely Amino Acid (AA) sequences. This guide assumes familiarity with general (nucleotide!) Augur workflows.
This guide first explains the changes needed to analyse a set of (unaligned) AA sequences representing a single gene/CDS, including ancestral reconstruction. We then describe how you can concatenate multiple genes together for increased resolution. Finally there’s a summary of the differences in Auspice behaviour when viewing protein-only datasets.
Note
For deeply diverged proteins you should use other approaches, and pay particular attention to model selection. Our temporal inference & tree refinement approach is not well suited to very long branches.
Prerequisites
Metadata TSV
AA sequence FASTA (unaligned or aligned)
Reference sequence for alignment, if not included in your sequences. FASTA or GenBank format.
Changes compared to a nucleotide workflow
Indexing, Filtering, and Subsampling
These commands now handle AA sequences when used with the --seq-type aa argument.
Note that the index file format is different for AA vs nuc sequences.
Alignment
augur align --method mafft (the default) will work for protein alignments.
Note that Nextclade doesn’t support AA inputs and thus can’t be used as an aligner in this case.
Tree construction
IQ-TREE can be parameterized for AA sequences by adding the the following arguments to
augur tree:
--method iqtree(the default)--tree-builder-args '--seqtype AA'--substitution-model <MODEL>The default IQ-TREE substitution model isLG; see the “Specifying substitution models” section of the IQ-TREE docs for a full list of available models.
An example command to construct a tree from a concatenated HA1 + HA2 alignment may look like:
augur tree \
--method iqtree \
--substitution-model LG \
--tree-builder-args '--seqtype AA' \
--alignment results/HA12/aligned.fasta \
--output results/HA12/tree_raw.nwk
Temporal inference (refine)
Augur refine will handle AA alignments if provided with --seq-type aa.
Internally this changes TreeTime to use the Jones-Taylor-Thornton (JTT92)
substitution model.
You may also want to use --branch-length-inference marginal depending on the
branch lengths involved, which better handles uncertainty in long branches.
An example command may look like:
augur refine \
--seq-type aa \
--alignment results/HA12/aligned.fasta \
--metadata results/metadata.tsv \
--tree results/HA12/tree_raw.nwk \
--timetree --root best --clock-filter-iqd 1 --date-confidence \
--branch-length-inference marginal \
--output-tree results/HA12/tree.nwk \
--output-node-data results/HA12/branch_lengths.json
Ancestral reconstruction
For nucleotide analyses there are two main approaches to reconstructing
amino-acid sequences and thus inferring AA mutations across the tree: augur
ancestral which independently translates provided nuc & AA alignments, and
augur translate which translates nucleotide sequences reconstructed across
the tree.
augur ancestral can work for AA-only analyses. You will need to provide the following options to reconstruct a single gene/CDS:
--genes <GENE NAME>--translations <FILENAME>(amino acid FASTA) the filename can be a specific path or include a%GENEplaceholder which will be replaced with the gene name[optional]
--root-sequence <FILENAME>(amino acid FASTA) the filename can be a specific path or include a%GENEplaceholder which will be replaced with the gene name[optional]
--annotation <GenBank or GFF>(not needed for a single gene analysis)
See Understanding root and reference sequences for more information about how providing a root-sequence will assign mutations to the root-node of the tree.
The optional annotation file allows the actual nucleotide coordinates of the gene to be used in the resulting JSON, which slightly changes how the dataset will appear in Auspice (see below).
An example command, where we constructed the tree from a concatenated HA1 + HA2
alignment but where we want to reconstruct mutations on HA1 and HA2 alignments
separately (results/HA1/aligned.fasta, results/HA2/aligned.fasta) would
look like:
augur ancestral \
--tree results/HA12/tree.nwk \
--annotation config/genemap.gff \
--translations results/%GENE/aligned.fasta \
--genes HA1 HA2 \
--output-node-data results/HA12/muts.json
Other commands (export, traits etc)
Other Augur commands in a typical workflow should work with protein sequences, including:
augur traitsaugur export v2
Notably, augur proximity (and the associated subsampling configs) do not yet work with AA sequences.
Concatenating multiple genes together for increased resolution
For increased resolution you may wish to concatenate multiple genes/CDSs together, but still infer the ancestral sequences independently for each gene. The main difference from the workflow described above is the need for sequence concatenation, which can be done using Seqkit
rule combine_alignments:
input:
alignments = ["<PATH>", "<PATH>", ...]
output:
alignment = "<PATH>"
shell:
"""seqkit concat {input.alignments} > {output.alignment}"""
Tree building and temporal inference should use this concatenated alignment.
For reconstruction of ancestral AA sequences you will want to reconstruct
each individual CDS. This will require a GenBank or GFF file which describe the
coordinates of the --genes you provide, and which must have nucleotide
annotations defined even thought they are unused. The (aligned) FASTA files for
each gene should have a filename which can be referred to using a %GENE
placeholder, and if you want to supply an AA root sequence for each gene they
will also need this pattern (supplying root sequences allows annotation of
mutations on the root node of the tree representing differences between the root
node and the supplied reference). An example invocation:
augur ancestral \
--tree tree.nwk \
--annotation HA_genemap.gff \
--genes HA1 HA2 \
--translations alignment_%GENE.fasta \
--root-sequence root-sequence_%GENE.fasta \
--output-node-data muts.json
Visualisation in Auspice
For the most part protein-only analyses work in Auspice as expected. The tree divergence (branch lengths) will be expressed either in units of amino acid mutations or amino acid substitutions per codon site, and of course there won’t be any nucleotide mutations annotated on branches!
Single gene datasets
For analyses which reconstruct mutations (augur ancestral) for a single
gene, the entropy panel will be limited to viewing that gene (Figure 1). The
entropy view uses two axes, with the upper one showing AA-coordinates (relative
to the gene) and the lower showing the corresponding nucleotide positions. The
nucleotide coordinate system used depends on whether an annotation file
(genemap) was provided to augur ancestral (Figure 1).
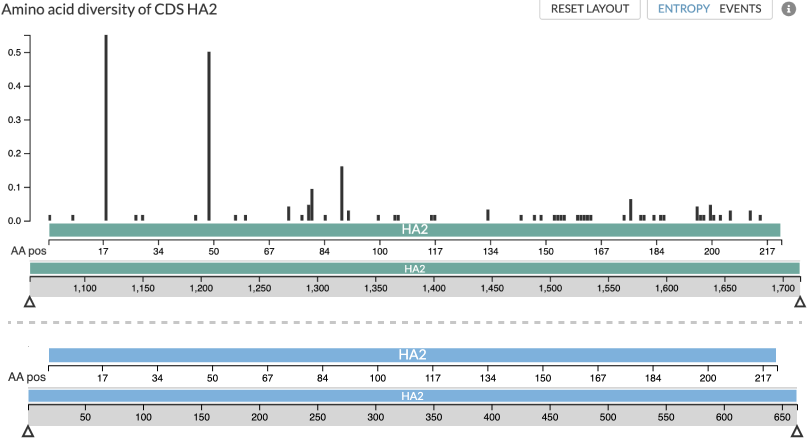
Figure 1: Two screenshots of how a single gene analysis may be displayed in Auspice. The teal screenshot is from an analysis using a genemap so that the nucleotide axis (the lower of the two teal axes) uses the true nucleotide coordinates of HA2: 1053..1715nt. The blue screenshot is from an analysis without use of a genemap, and thus the nucleotide coordinates are simply placeholders for AA sequence: 1..3*len(aa). In both cases the upper axis represents the same AA-coordinates of the gene/gene-fragment.
For analyses which reconstruct multiple genes, such as in the code snippets above, the entropy panel will use a placeholder nucleotide genome view in order to allow navigation between the different genes (Figure 2). This genome will span the nucleotide positions of the genes, and clicking on them will display that gene in AA-space, similar to that in Figure 1 above.
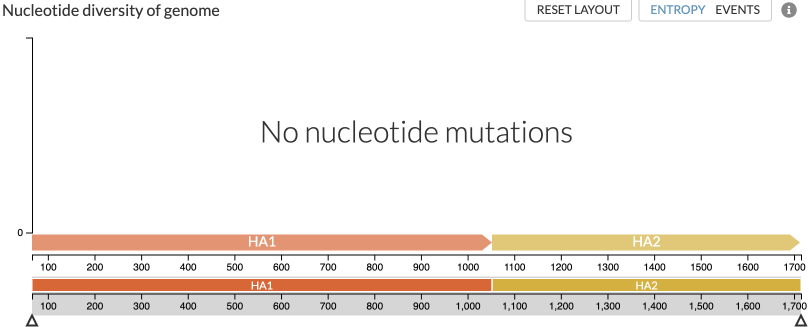
Figure 2: View of a multi gene analysis which starts using a placeholder nucleotide view, allowing you to navigate to the available genes (HA1 and HA2 in this case). There are no nucleotide mutations in this placeholder view, but selecting (clicking on) HA1 or HA2 will show the AA-diversity of that peptide, similar to Figure 1.